Investigational Brochure Fda
Investigational Brochure Fda - 29028) the sponsor is conducting a phase 1. The investigator’s brochure (ib) is a comprehensive compilation of clinical and nonclinical data on the investigational product (drug, supplement, device or other product) maintained by a drug. Implementation of the regimen will begin immediately for investigational new drug (ind) applications, where inclusion of nams data is encouraged, and is outlined in a roadmap. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. (a) a sponsor who intends to conduct a clinical investigation subject to this part shall submit an “investigational new drug application” (ind) including, in the following order: The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. Good clinical practice (gcp) is an international ethical and scientific. Ind content and format for phase 1 studies. Fda regulations [21 cfr 312.23 (a)(5)] state that an investigator's brochure must contain the following information: Studies using a drug that has not been approved by the food and drug administration (fda) or for indications not in the approved labeling may require filing an investigational new drug. The psp documents the investigator’s acknowledgment of receipt, their review of the protocol, and their agreement to conduct the study according to its terms. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. The first investigational new drug (ind) application for sbt777101 has been approved in rheumatoid arthritis (ra ). Implementation of the regimen will begin immediately for investigational new drug (ind) applications, where inclusion of nams data is encouraged, and is outlined in a roadmap. Ind application sponsors are expected to submit brief reports of the progress of the investigations conducted under their respective ind application within 60 days of the anniversary date that. A brief description of the drug substance and the formulation, including. Good clinical practice (gcp) is an international ethical and scientific. Fda regulations [21 cfr 312.23 (a)(5)] state that an investigator's brochure must contain the following information: The investigator’s brochure (ib) is a comprehensive compilation of clinical and nonclinical data on the investigational product (drug, supplement, device or other product) maintained by a drug. Providing investigators with the necessary information to. Studies using a drug that has not been approved by the food and drug administration (fda) or for indications not in the approved labeling may require filing an investigational new drug. (a) this part contains procedures and requirements governing the use of investigational new drugs, including procedures and requirements for the submission to, and review by, the food. Fda regulations. Ind application sponsors are expected to submit brief reports of the progress of the investigations conducted under their respective ind application within 60 days of the anniversary date that. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. Fda regulations. The psp documents the investigator’s acknowledgment of receipt, their review of the protocol, and their agreement to conduct the study according to its terms. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. In drug development, the investigator’s brochure (ib). To discuss an alternative approach, contact the fda office responsible for this guidance as listed on the title page. Identification, quality, purity, and strength of the investigational drug varies with the phase of the investigation “…fda's review of phase 1 submissions will focus on assessing The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the. The investigator’s brochure (ib) is a comprehensive compilation of clinical and nonclinical data on the investigational product (drug, supplement, device or other product) maintained by a drug. Implementation of the regimen will begin immediately for investigational new drug (ind) applications, where inclusion of nams data is encouraged, and is outlined in a roadmap. (a) a sponsor who intends to conduct. The psp documents the investigator’s acknowledgment of receipt, their review of the protocol, and their agreement to conduct the study according to its terms. Studies using a drug that has not been approved by the food and drug administration (fda) or for indications not in the approved labeling may require filing an investigational new drug. Identification, quality, purity, and strength. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. The first investigational new drug (ind) application for sbt777101 has been approved in rheumatoid arthritis (ra ). 29028) the sponsor is conducting a phase 1. Providing investigators with the necessary information to. Ind content and format. (a) this part contains procedures and requirements governing the use of investigational new drugs, including procedures and requirements for the submission to, and review by, the food. The first investigational new drug (ind) application for sbt777101 has been approved in rheumatoid arthritis (ra ). 29028) the sponsor is conducting a phase 1. In drug development, the investigator’s brochure (ib) summarises. The investigator’s brochure (ib) is a comprehensive compilation of clinical and nonclinical data on the investigational product (drug, supplement, device or other product) maintained by a drug. Identification, quality, purity, and strength of the investigational drug varies with the phase of the investigation “…fda's review of phase 1 submissions will focus on assessing Ind content and format for phase 1. Ind content and format for phase 1 studies. Ind application sponsors are expected to submit brief reports of the progress of the investigations conducted under their respective ind application within 60 days of the anniversary date that. 29028) the sponsor is conducting a phase 1. To discuss an alternative approach, contact the fda office responsible for this guidance as listed. A brief description of the drug substance and the formulation, including. Providing investigators with the necessary information to. To discuss an alternative approach, contact the fda office responsible for this guidance as listed on the title page. (a) a sponsor who intends to conduct a clinical investigation subject to this part shall submit an “investigational new drug application” (ind) including, in the following order: Good clinical practice (gcp) is an international ethical and scientific. The psp documents the investigator’s acknowledgment of receipt, their review of the protocol, and their agreement to conduct the study according to its terms. The first investigational new drug (ind) application for sbt777101 has been approved in rheumatoid arthritis (ra ). Background clinical study reports (csrs) are standardized full reports of the protocols, results, and other pertinent details of clinical studies that are typically submitted by. Investigational new drug (ind)is an application that is submitted to us fda by a pharmaceutical company to obtain permission from the agency to start human clinical Ind content and format for phase 1 studies. 29028) the sponsor is conducting a phase 1. The investigator’s brochure (ib) is a comprehensive compilation of clinical and nonclinical data on the investigational product (drug, supplement, device or other product) maintained by a drug. Ind application sponsors are expected to submit brief reports of the progress of the investigations conducted under their respective ind application within 60 days of the anniversary date that. Studies using a drug that has not been approved by the food and drug administration (fda) or for indications not in the approved labeling may require filing an investigational new drug. Fda regulations [21 cfr 312.23 (a)(5)] state that an investigator's brochure must contain the following information: The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects.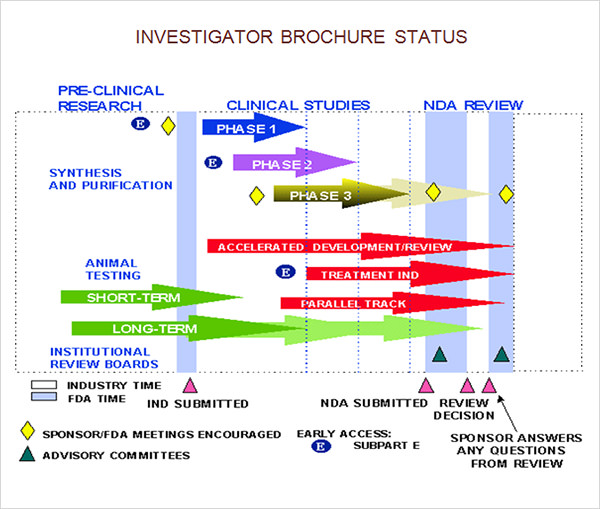
8+ Investigator Brochures Sample Templates

Investigator Brochure Template Fda
Investigators Brochure Pharmacology

Investigator Brochure Template Fda

Investigator Brochure Template Fda

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

PPT What Is An IND? PowerPoint Presentation, free download ID263381

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word
Identification, Quality, Purity, And Strength Of The Investigational Drug Varies With The Phase Of The Investigation “…Fda's Review Of Phase 1 Submissions Will Focus On Assessing
The Investigator’s Brochure (Ib) Is A Compilation Of The Clinical And Nonclinical Data On The Investigational Product(S)1 That Are Relevant To The Study Of The Product(S) In Human Participants.
Implementation Of The Regimen Will Begin Immediately For Investigational New Drug (Ind) Applications, Where Inclusion Of Nams Data Is Encouraged, And Is Outlined In A Roadmap.
High Quality Protocols Facilitate Proper Planning, Conduct, Reporting, And External Review Of Randomised Trials, Yet Their Completeness Varies And Key Elements Are Often Not.
Related Post:
